
If an applicant wishes to obtain a license in one Member State (MS) an application must be made to the national Competent Authority (CA) which then issues a national license. Since 1998 national licenses are strictly limited to the initial phase of Mutual Recognition.
Regulation (EC) No 1084/2003 (MRP or DCP) and Regulation (EC) No 1085/2003 (for CP) describe in annex II the types of applications which fall outside the scope of “variations” but at the same time are not complete new applications. Annex II lists three main categories for existing licensed products with:
- Changes to the active substance
- Changes to strength, pharmaceutical form, and route of administration
- Other changes specific to veterinary medicinal products to be administered to food producing animals
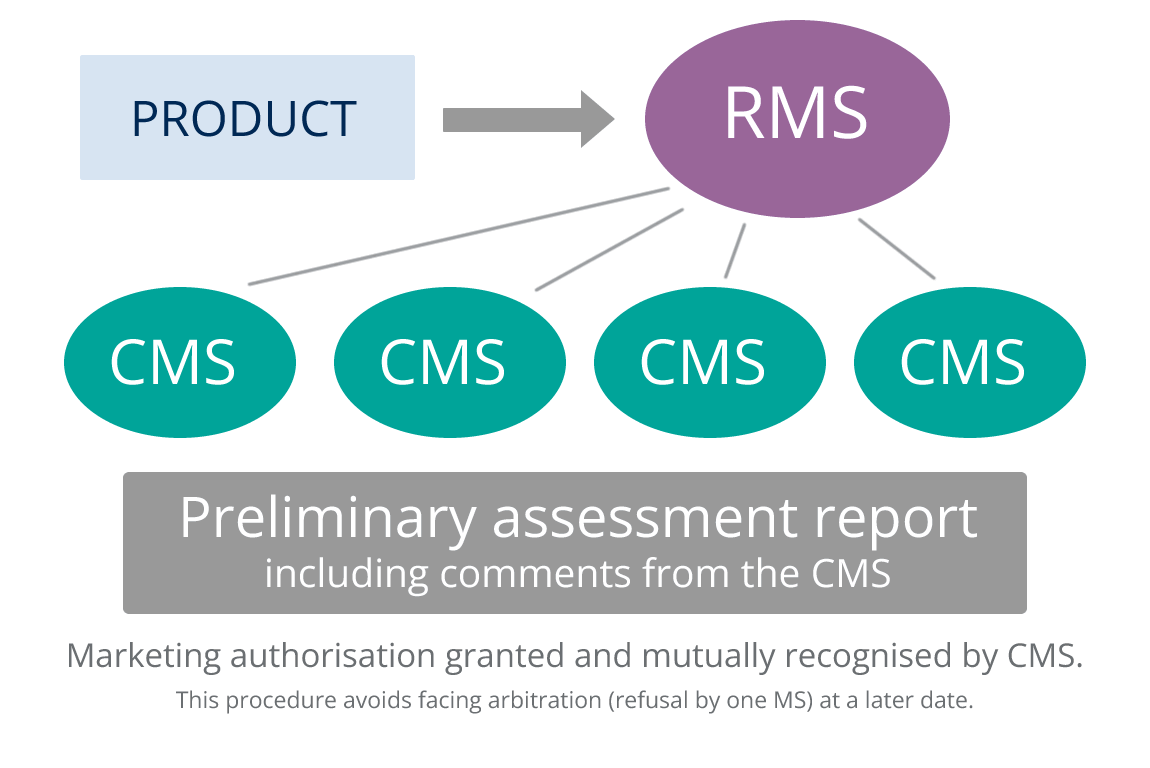
For products to be registered in more that one Member State (MS) and which do not qualify for the Centralised Procedure (CP) applicants must use either the Decentralised Procedure (DCP) or the Mutual Recognition Procedure (MRP). The MRP is to be used if the aim is to register in more than one Member State and the medicinal product in question has already received a Marketing Authorisation (MA) in any MS at the time of application.
The MRP is based on the idea that a national license approved in one EU Member State (Reference Member State – RMS) should be mutually recognised in other EU countries (Concerned Member States – CMS). This is based on the assumption that the evaluation criteria in the EU member states are sufficiently harmonised and are of the same standard. At the end of a MRP national licenses are issued in the CMSs involved in the procedure.
Harmonisation throughout the life cycle of the product is ensured by the application of variations (detailed in Regulation (EC) No 1084/2003) simultaneously in the RMS and the CMSs. To this end the Marketing Authorisation Holder (MAH) must ensure that they have the resources to achieve this concurrent submissions in all countries involved in the MRP.
In order to obtain a Community Authorization, an application is made to the EMEA – The European Medicines Agency. The application is scientifically evaluated by the Committee for Medicinal Products for Human Use (CHMP) or the Committee for Medicinal Products for Veterinary Use (CVMP). The European Commission receives an opinion within 210 days and drafts a decision on a Community MA. A MA granted under the CP is valid for the entire EU market.
The Centralised Procedure (CP) is mandated for:
- Biotechnology products
- Orphan medicinal products and
- Medicinal products containing a new active substance intended to treat acquired immune deficiency syndrome (AIDS), cancer, neurodegenerative disorders and diabetes
- Therapeutically innovative products
- OTC products with a clear added value for the community
- Certain generic products